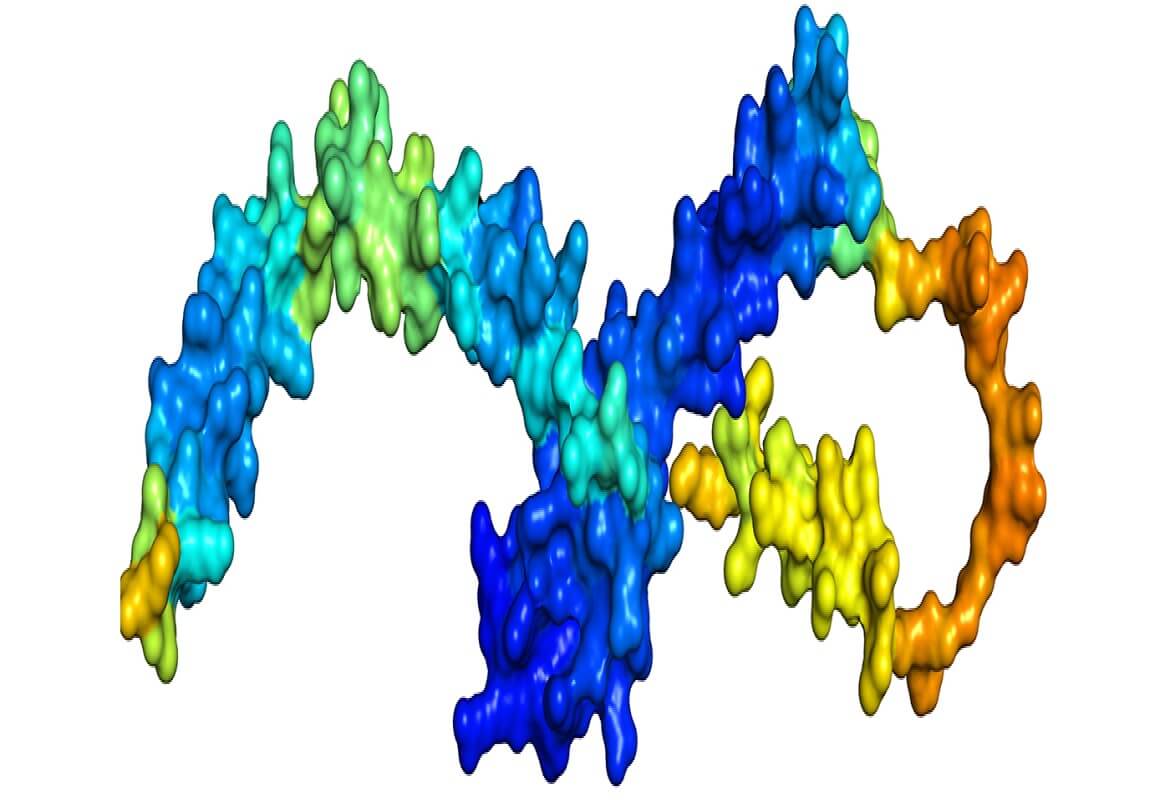
人工智能預測蛋白質結構和相互作用
此時此刻,生物學的根基似乎正在動搖。近幾個月來,大量論文突破了人工智能(AI)在蛋白質結構和蛋白質-蛋白質相互作用預測方面的極限。
2020 年,DeepMind 團隊從倫敦國王十字附近的黑鋼建築出發,前往參加第 14 屆年度 CASP(蛋白質結構預測技術批判性評估)競賽。他們的模型打破了所有預期,GDT_TS(基本上是與“真實”模型相比正確預測的蛋白質比例)達到了 92.4。結果於次年發表在《自然》雜誌上。
看到結果後,該領域的許多人稱蛋白質折疊問題“已解決”。哥倫比亞大學的系統生物學家 Mohammed AlQuraishi 撰寫了一篇關於 AlphaFold 成就的博客;“感覺就像一個孩子離開了家。” 2
CASP14 剛剛過去兩年。從那時起,一批名副其實的研究人員紛紛跳上人工智能的列車,運用自己的想法,在飛速發展的旋風中擴展該技術的潛力。
更好的結構
CASP14 是人工智能蛋白質結構預測的分水嶺,但真正的革命早在幾年前就開始了。芝加哥豐田技術研究所教授 Jinbo Xu 在 2016 年使用卷積神經網絡(一種深度學習模型)來預測數百種蛋白質的三級結構。3當時,“對於單一蛋白質結構預測來說,這一改進確實非常重要,”他說。“在 CASP13 中,這種深度學習模型將預測質量從不到 40 分提高到了 60 分以上。”
2020 年初,第一個 AlphaFold 模型發表在Nature上。它也使用神經網絡來預測結構,對 43 個蛋白質結構域實現了 0.7 或更高的準確度。4大約在同一時間,華盛頓大學的 David Baker 實驗室發布了他們自己的模型 RoseTTAfold,該模型可以快速生成“僅根據序列信息的準確的蛋白質-蛋白質複合物模型”。5
從一般的結果轉向驚人的準確性(例如使用 AlphaFold2 實現的結果)並不容易。即使在今天,大多數預測模型也依賴於兩大類數據:蛋白質序列和蛋白質結構。徐說,如果該蛋白質“在數據庫中”“沒有任何序列同源物”或類似的保守蛋白質,那麼高精度預測天然蛋白質的結構仍然具有挑戰性。然而,2021 年,Xu 及其同事在《自然·機器智能》雜誌上表明,僅使用序列數據的大型神經網絡就可以預測一半以上“硬測試”蛋白質的結構,精度超過 80%,且無需依賴共同進化信息。6
如今,AlphaFold2 仍然是結構預測的主導模型,但其他團隊已經擴展了這項工作。該模型現在通常用於“產生幻覺”或想像出具有自然界中未發現的功能的全新蛋白質。在最近的一項研究中,研究人員開發了一個大型語言模型(很像文本到圖像工具 DALL-E)來生成從頭蛋白質。一種名為 ProteinMPNN 的軟件工具可以在大約一秒鐘內完成此操作,無需任何專家培訓。7通過將該工具與 AlphaFold 結合使用,研究人員可以快速生成蛋白質、模擬其結構並改進方法來尋找具有所需特性的蛋白質。
從很多方面來說,這一成就標誌著生物學人工智能新時代的黎明:使用算法不是為了解決現有蛋白質的結構,而是為了夢想全新的可能性。
接觸蛋白質
人類基因組包含大約 20,000 個蛋白質編碼基因。人們認為,在任何給定的細胞中都有超過十萬種獨特的蛋白質-蛋白質相互作用(PPI)。8繪製這種複雜性是生物學中的一項巨大挑戰,也是人工智能工具獨特適合的另一個問題。
不幸的是,預測 PPI 比預測單一蛋白質的結合更具挑戰性,麻省理工學院博士後 Felix Wong 說。“即使是一個小分子也可能有幾十個原子,弄清楚它撞擊蛋白質的位置很複雜,”他補充道。“單個蛋白質中可能有數十個結合袋。”
AlphaFold 目前僅預測蛋白質結構的單個快照。但是,在活細胞內,當蛋白質與其他蛋白質接觸時,它們會扭曲和彎曲。那麼,更有用的預測工具將生成一系列潛在結構。
儘管如此,許多團隊已經將人工智能模型專門應用於蛋白質相互作用和復合物。去年,AlphaFold 發布了多鏈蛋白質複合物的 Multimer 模型。9與基線 AlphaFold 方法相比,它取得了相當大的改進。它的工作原理如下:首先,為複合體構建多序列比對以推斷進化關係。然後,使用與 AlphaFold2 基本相同的深度學習方法來預測三級結構。一個名為 AlphaPulldown 的開源 Python 包也可用於快速運行 AlphaFold-Multimer 模型。10
例如,類似的蛋白質複合物模型已用於研究大腸桿菌蛋白質組,並破譯具有挑戰性的蛋白質簇的結構,包括細胞色素 c 生物發生系統中的蛋白質簇。11
隨著 PPI 的人工智能模型不斷改進,徐打算使用它們來進行“虛擬藥物”篩選。“如果我們有一個非常好的算法來預測[蛋白質-蛋白質相互作用],”他說,“那麼我們就可以對抗體進行虛擬篩選。” 然而,Wong 和麻省理工學院的另一位博士後研究員 Aarti Krishnan 最近的一項研究表明,基於 AlphaFold 的對接模型目前無法準確預測蛋白質與抗生素的相互作用,儘管抗生素是比抗體原子少得多的小分子。12
儘管如此,有希望的進展即將到來。AI模型可用於直接改進對接工具,例如AutoDock、DOCK、LeDock或FlexAID,以快速篩選針對蛋白質的小分子。最近有一個名為 EquiBind 的工具使用有關蛋白質幾何形狀的特定假設,並將其與機器學習模型結合起來,以加速藥物-蛋白質相互作用測試,13 這可能有望帶來更深入的學習引導方法進行對接 。
克里希南說,未來進步的一個關鍵限制是數據。“我們希望使用實驗訓練數據集來改進機器學習模型,”她解釋道。例如,擁有更多用於與配體複合的蛋白質的冷凍電子顯微鏡結構將特別有用,這可能會改善目前主要在分離蛋白質上訓練的模型的訓練數據集。
文章作者-biocompare